“Senescent cells release a senescence-associated secretory phenotype (SASP),including exosomes that may act as signal transducers between distal tissues, and propagate secondary senescence.”
As the global population grows older, understanding what drives the aging process is becoming increasingly important. Diseases like Alzheimer’s, cardiovascular conditions, and cancer are more common with age, yet many current treatments only manage symptoms rather than addressing the underlying biological causes.
One contributor to aging is the buildup of “senescent” cells—cells that have stopped dividing but do not die. These cells can harm nearby tissues by releasing molecular signals, a process known as secondary senescence.
The team led by Sandip Kumar Patel, Joanna Bons, and Birgit Schilling from The Buck Institute for Research on Aging focused their study on exosomes—tiny, bubble-like structures released by cells that carry proteins, lipids, and genetic material. These particles can move through the bloodstream and influence distant tissues.
The researchers wanted to know whether exosomes from senescent cells and from the blood of older adults shared common markers of aging. Since aging cells are spread throughout the body and lack a single clear marker, exosomes could provide a new way to detect their presence through a simple blood test.
To explore this, the team analyzed exosomes from two sources: lab-grown human lung cells that had undergone senescence and blood samples from both young (20–26 years old) and older (65–74 years old) adults. They used high-throughput mass spectrometry.
Results: Exosomes Reveal Signs of Aging
In total, the team identified over 1,300 proteins and 247 lipids within the exosomes. Specifically, 52 proteins appeared in both senescent cells and the blood plasma of older adults, many of which are associated with inflammation and tissue damage. Some examples include Prothrombin, Plasminogen, and Reelin—molecules involved in blood clotting, tissue remodeling, and neural development. Their presence in both aged blood and senescent cells suggest a broader impact of aging on multiple biological systems.
The team also observed significant changes in the lipid content of the exosomes. Lipids that help maintain cell membrane structure were more common in samples from older individuals, while lipids involved in energy storage were less abundant.
In addition, the researchers detected changes in microRNAs—small pieces of genetic material that regulate gene expression. Several microRNAs found in the blood of older adults have already been associated with diseases such as Alzheimer’s and osteoarthritis.
The Impact: Potential for Diagnostics and Anti-Aging Therapies
This study is among the first to directly compare exosomes from senescent cells and human plasma, revealing shared aging-related markers across biological systems.
These particles act like messengers, spreading signals that may accelerate aging in other cells. This supports the concept of secondary senescence—where aging-like behavior is transmitted from senescent cells to healthy ones—suggesting that exosomes may help propagate aging throughout tissues over time.
This work could lead to the development of blood tests that measure biological age more accurately than a person’s chronological age. It might also help clinicians monitor the effectiveness of anti-aging treatments.
Future Perspectives and Conclusion
Although the study involved a small number of human samples, it presents a promising new approach to studying aging. If confirmed in larger studies, the findings could lead to improved diagnostic tools and therapies for age-related diseases.
In the long term, researchers may explore ways to block or modify harmful exosome signals to protect healthy cells from premature aging. These molecular signatures could also support personalized medicine approaches or help track the effectiveness of anti-aging interventions in clinical settings.
Click here to read the full research paper published in Aging (Aging-US).
___
Aging is indexed by PubMed/Medline (abbreviated as “Aging (Albany NY)”), PubMed Central, Web of Science: Science Citation Index Expanded (abbreviated as “Aging‐US” and listed in the Cell Biology and Geriatrics & Gerontology categories), Scopus (abbreviated as “Aging” and listed in the Cell Biology and Aging categories), Biological Abstracts, BIOSIS Previews, EMBASE, META (Chan Zuckerberg Initiative) (2018-2022), and Dimensions (Digital Science).
Click here to subscribe to Aging publication updates.
“This collection is published in memory of Professor Judith Campisi, a pioneering force in the field of cellular senescence whose groundbreaking work shaped the understanding of senescence in aging, cancer, and tissue homeostasis.”
BUFFALO, NY — May 1, 2025 —Aging (Aging-US) invites submissions for a Special Collection dedicated to the theme of cellular senescence, spanning its basic mechanisms, physiological and pathological functions, and clinical applications.
This collection is published in memory of Professor Judith Campisi, a pioneering force in the field of cellular senescence whose groundbreaking work shaped the understanding of senescence in aging, cancer, and tissue homeostasis. Her legacy continues to inspire generations of scientists working to decode the complex biology of senescent cells and their impact on health and disease.
We welcome original research articles, reviews, and perspectives on topics including:
Fundamental mechanisms of senescence induction and maintenance
Regulation and context-specific roles of the senescence-associated secretory phenotype (SASP)
Beneficial and detrimental effects of senescent cells in vivo
Senescence in development, aging, regeneration, and age-related diseases
Biomarkers, imaging, and tools for senescence detection and quantification
Therapeutic targeting of senescent cells: senolytics, senomorphics, and clinical translation
This Special Collection is guest edited by Han Li and Irina Conboy, both internationally recognized leaders in the study of senescence and aging.
Researchers evaluated three different mouse strains with varying sensitivity to radiation lung fibrosis in an effort to uncover the underlying mechanisms.
The Trending With Impact series highlights Aging publications (listed as “Aging (Albany NY)” by Medline/PubMed and “Aging-US” by Web of Science) that attract higher visibility among readers around the world online, in the news, and on social media—beyond normal readership levels. Look for future science news about the latest trending publications here, and at Aging-US.com.
—
Listen to an audio version of this article
Radiation is an effective treatment for many types of cancer. Unfortunately, this treatment has the potential to cause long-term side effects in some patients, including the thickening or scarring of lung tissue, known as pulmonary fibrosis. Radiation-induced pulmonary fibrosis (RIPF) is a serious complication that can occur after radiation therapy and can lead to death. Predicting an individual’s risk of developing RIPF remains challenging for clinicians, as little is known about the underlying mechanisms that cause it.
“Differential susceptibility to lung injury from radiation and other toxic insults across mouse strains is well described but poorly understood.”
While the precise mechanisms underlying RIPF are not fully understood, it is thought that senescent pneumocytes (or alveolar cells) play a key role. Pneumocytes are a type of cell in the lung that are essential for gas exchange. Type II pneumocytes (AECII) function as alveolar stem cells after lung injury. The researchers hypothesized that macrophages (a type of white blood cell that play an important role in immune responses) may contribute to promoting AECII senescence.
“AECII are known to be in close contact with alveolar macrophages, and, in this fashion, to contribute to lung homeostasis [11].”
The researchers hypothesized that natural variations in macrophage function contribute to differences in RIPF susceptibility. To explore their hypothesis, they evaluated three different mouse strains with varying sensitivity to radiation lung fibrosis: C57L mice (RIPF-prone), C57BL6/J mice (intermediate) and C3H/HeN mice (RIPF-resistant). Female mice (to avoid sex-based differences in results) underwent thoracic irradiation (IR). Changes in macrophages and pneumocytes were assessed.
The Results
The team found that susceptibility to radiation-induced lung injury and premature AECII senescence varied by mouse strain. Pulmonary irradiation led to varied macrophage phenotypes and accumulation in each strain. In responses to polarizing stimuli, macrophages demonstrated strain-dependent responses. M2 macrophages induced AECII senescence via NOX2-derived superoxide production in a strain-dependent manner. Finally, macrophages expressing NOX2 accumulated in fibrotic lungs after radiation.
“NOX1 and NOX2 protein were expressed at the highest levels in C57L BMDM, with intermediate expression in C57BL6/J BMDM and the lowest expression in C3H/HeN BMDM (Figure 6B).”
The researchers demonstrated that the C57L mice (the strain with the greatest sensitivity to RIPF) exhibited the greatest rate of accumulation of senescent AECII cells. At the same time, they found that the fibrosis-sensitive (C57L and C57Bl6/J) mouse strains exhibit a greater accumulation of M2 polarized macrophages than the fibrosis-resistant strain (C3H/HeN).
“However, until now, the impact of M2 polarization on AECII senescence was unexplored. In this study, we identified that M2 macrophage polarization can contribute to AECII senescence, potentially leading to a positive feedback loop that furthers pulmonary injury.”
Conclusion
This study provides new insights into the role of macrophages in RIPF susceptibility. The findings suggest that natural variations in macrophage function contribute to differences in RIPF susceptibility. The different macrophage polarization profiles across strains may contribute to their varying susceptibilities to RIPF by promoting AECII senescence. These findings may help to develop new strategies for the prevention and treatment of RIPF.
“In this study, variation in the accumulation of senescent cells across strains with varying sensitivity to fibrosis has been established. Further, strain variation in macrophage response to polarizing stimuli and capacity to produce superoxide and induce senescence in epithelial cells is described. Together, these data highlight the importance of macrophage-epithelial interactions in the context of lung fibrosis and identify NOX2 as a possible therapeutic target in radiation lung injury.”
Click here to read the full research paper published by Aging.
Aging is an open-access journal that publishes research papers bi-monthly in all fields of aging research. These papers are available at no cost to readers on Aging-us.com. Open-access journals have the power to benefit humanity from the inside out by rapidly disseminating information that may be freely shared with researchers, colleagues, family, and friends around the world.
Published on the cover of Aging’s Volume 14, Issue 7, researchers conducted a new study investigating the role of IGFBP5 in replicative senescence.
The Trending With Impact series highlights Aging (Aging-US) publications that attract higher visibility among readers around the world online, in the news, and on social media—beyond normal readership levels. Look for future science news about the latest trending publications here, and at Aging-US.com.
—
Listen to an audio version of this article
In 1961, Leonard Hayflick and Paul Moorhead proposed a theory later named the Hayflick Limit. They discovered that a normal human cell can divide between 50 and 70 times before it can no longer proliferate and eventually dies. Researchers have since continued to explore this phenomenon and, today, this aging process is known as cellular (replicative) senescence.
“There are currently several experimental models of cellular senescence. Hayflick and Moorhead observed that primary human fibroblasts in culture exhibit a limited proliferative capacity [6]. This growth arrest during passages is called replicative senescence.”
This permanent cessation of the cell cycle is universally found in biology due to known and unknown causes, including the shortening of telomeres. While telomere shortening plays an important role, it is not the only event responsible for inducing cellular senescence. Thus, researchers have spent decades under the microscope experimenting with cellular models of replicative senescence.
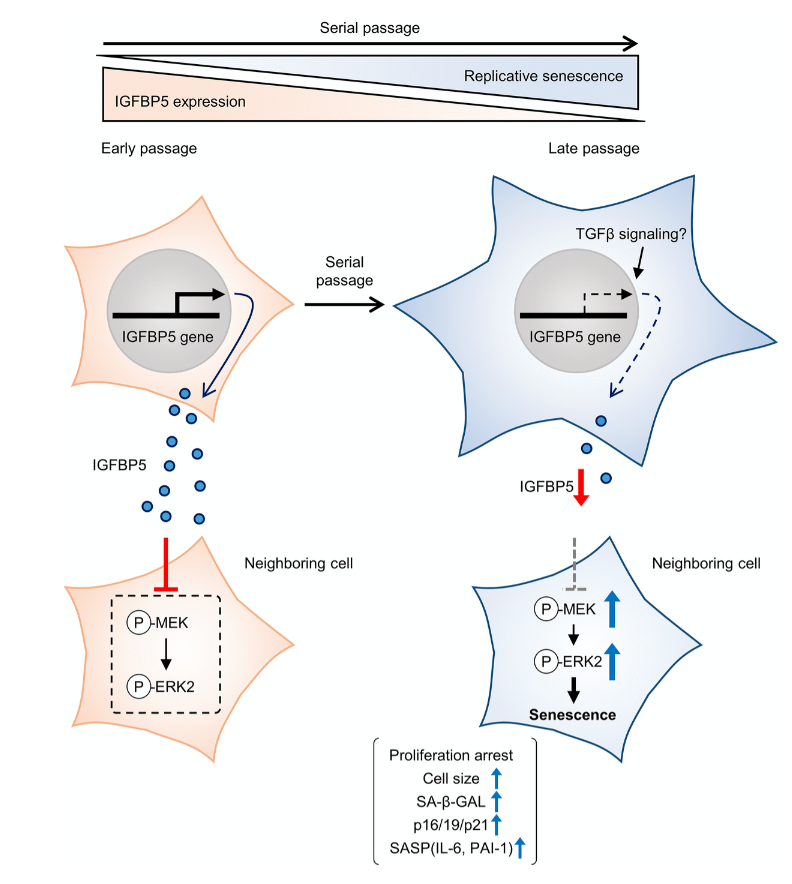
Cellular senescence is typically characterized by cell growth arrest, an increase of cells positive for SA-β -gal staining, and upregulation of p16 and p19. To begin this study, the team cultured embryonic mouse fibroblasts (MEFs) and conducted cell passages according to the 3T3 method. They found that the MEFs underwent senescence after the 5th passage (P5). The team also found that at P8, the expression of insulin-like growth factor binding protein 5 (IGFBP5) mRNA was significantly reduced when compared with that of P2 MEFs.
Next, the team performed a knockdown of IGFBP5 in the MEF cells. Results showed that IGFBP5 knockdown induced premature cellular senescence in P2 MEFs. Knockdown of IGFBP5 increased phosphorylation of extracellular signal-regulated kinases 1 and 2 (ERK1/2) but did not affect expression levels of Akt or p16 repressors. The researchers also found that supplementing the cell culture growth medium with additional exogenous IGFBP5 delayed growth arrest and reduced replicative senescence in the MEF cells.
“To examine whether activated ERK1 and ERK2 by IGFBP5 knockdown are involved in the induction of senescent phenotypes, we examined effects of knockdown of ERK1 and ERK2 using a combination with IGFBP5 siRNA in P2 MEFs.”
Upon further analysis of ERK1/2’s role in IGFBP5-knockdown cells, the team found that the silencing of ERK2, and not ERK1, blocked the increase in the number of SA-β-GAL-positive cells. ERK2 knockdown attenuated the reduction in the cell number and upregulation of p16 and p21 in IGFBP5-knockdown cells. This study provides evidence that downregulation of IGFBP5 contributes to replicative senescence via ERK2 activation in mouse embryonic fibroblasts.
Conclusion
For the first time, the role of IGFBP5 in replicative senescence was demonstrated in MEFs. Their findings suggest that ERK2 underlies cellular senescence induced by IGFBP5 downregulation. Cellular senescence appears to be a complex process with many moving parts. While more research is needed to fully understand the role of IGFBP5 in replicative senescence, this study provides new insights into the underlying mechanisms involved in this complex process.
“In conclusion, the results of the present study demonstrated that downregulation of IGFBP5 during serial passage contributes to replicative senescence via an ERK2-dependent mechanism (Figure 6). The results suggest that IGFBP5 counteracts replicative senescence in MEFs.”
Figure 6. Schematic summary of our findings. MEFs at early passage secrete certain levels of IGFBP5. Secreted IGFBP5 proteins inhibit MEK/ERK2 by attenuating their phosphorylation (P) in the neighboring cell, leading to suppression of cellular senescence. IGFBP5 secretion is decreased during serial passage, causing activation of ERK2 and cellular senescence.
Click here to read the full research paper published by Aging (Aging-US).
Aging (Aging-US) is an open-access journal that publishes research papers bi-monthly in all fields of aging research. These papers are available at no cost to readers on Aging-us.com. Open-access journals have the power to benefit humanity from the inside out by rapidly disseminating information that may be freely shared with researchers, colleagues, family, and friends around the world.
In the cover paper of Aging (Aging-US) Volume 14, Issue 2, researchers discovered a potential therapeutic strategy to target senescent cells and combat aging and age-related diseases.
The Trending With Impact series highlights Aging (Aging-US) publications that attract higher visibility among readers around the world online, in the news, and on social media—beyond normal readership levels. Look for future science news about the latest trending publications here, and at Aging-US.com.
—
Listen to an audio version of this article
Cellular senescence appears to be a phenomenon fundamentally ingrained within the aging process and linked to age-related diseases. Characterized broadly by permanent cessation of the cell cycle, cellular senescence may not be as permanent as once thought.
Adenosine triphosphate (ATP) is an energy-carrying molecule found in all living cells. In order to meet the energy demands of the cell, the primary function of the mitochondria is to produce ATP. The maintenance of mitochondrial metabolism is inseparably linked with the regulation of senescence. Therefore, dysfunctional mitochondria has been considered as both a target and the cause of senescence. In addition to a marked decrease in ATP production, senescent cells also increase the expression of inflammatory cytokines, including interleukin 33, or IL-33. The researchers believe that reducing IL-33 may be a possible intervention to reduce senescence in aging patients and age-related diseases.
“In this study, using in-house compound library containing 20 oxazoloquinoline analogs designed to IL-33 inhibitors [9], we aimed to identify compounds capable of ameliorating senescence.”
The researchers investigated 20 oxazoloquinoline analogs using in vitro assays of senescent human diploid fibroblasts and embryonic kidney cells. Efficacy of the candidate compounds was determined using a screening strategy to measure their capacity to increase cell number. Cell numbers were measured between zero and 20 days after compound exposure. The researchers also measured indicators including mitochondrial membrane potential, reactive oxygen species (ROS) levels and p21 expression. They found that the analog KB1541 led to the maximum cell number increase, the recovery of mitochondrial function and the alleviation of cellular senescence. The researchers suggest that KB1541 could be a promising therapeutic agent for use in aging-related diseases.
“The increase in mitochondrial cristae length by KB1541 could be explained by previous findings showing that the increase in ATP generation exerted beneficial effects in mitochondrial function including increases in calcium buffering capacity and decrease in overall ROS production [48].”
CONCLUSIONS
“Taken together, our study provides evidence that the fine-tuning of ATP synthase 5 alpha/beta dimerization by KB1541 can induce mitochondrial functional recovery, concomitant recovery of senescent phenotypes, rendering the use of KB1541 as a potentially advantageous therapeutic strategy in aging and age-related diseases.”
The authors acknowledged that further studies are needed to clarify the exact relationship between IL-33 and mitochondrial energy metabolism. Further studies are also needed to investigate whether other IL-33 inhibitors can modulate senescence by the mechanisms found in the study. This research provides valuable insight into the potential of oxazoloquinoline analogs as novel therapeutic agents for aging and age-related diseases. With further exploration, their findings could lead to new therapeutic strategies to combat aging.
“The role of IL-33 in senescence is not clearly elucidated, therefore discovery of a novel interacting partner will provide clues toward revealing its function.”
Click here to read the full research paper published by Aging (Aging-US).
Aging (Aging-US) is an open-access journal that publishes research papers bi-monthly in all fields of aging research. These papers are available to read at no cost to readers on Aging-us.com. Open-access journals offer information that has the potential to benefit our societies from the inside out and may be shared with friends, neighbors, colleagues, and other researchers, far and wide.
Researchers explain their studies that were published in Aging
Behind the Study is a series of transcribed videos from researchers elaborating on their recent oncology-focused studies published in Aging. A new Behind the Study is released each Monday. Visit the AgingYouTube channel for more insights from outstanding authors.
—
Greetings. My name is Andrei Gudkov. I am working in Roswell Park Cancer Institute, designated cancer center located in Buffalo, New York. I am Senior Vice President for Basic Science and chair of Department of Cell Stress Biology. My research is focused on understanding of the mechanisms of deregulation of a variety of stress response pathways in cancer cells as well as in normal cells in relation to cancer origin, progression, or engraftment and trying to use the information which we are generating during this research to come up with new types of treatment of cancer or cancer prevention.
Recently, our interests have significantly switched towards studying of the mechanisms of aging in its relations to cancer, since, as we all know, both conditions are closely connected. During the last, probably 20 years, one of the central theories of aging in mammals has been evolving towards connection between chronic sterile inflammation, which is accumulating in tissues with age of a mammal, including humans, with systemic decline in regeneration capabilities, in function of organs and tissues, and increasing risk of major diseases, altogether known as aging-related diseases. And the source of this inflammation, its origin, has been the central focus of studies of many.
During last couple of years, the dominating opinion in the field is about the central role of senescent cells, cells which chose to stay irreversibly growth-arrested in response to DNA damage, which they acquire during their life. And, through that, change their phenotype in more significant way than just growth arrest, acquiring the ability to secrete a spectrum of pro-inflammatory factors.
These senescent cells, which initially were defined as such in tissue culture experiments, eventually were proclaimed to be the main suspects in their putative role of inflammation creators in aging organism. This idea has become really popular, especially following a series of brilliant works coming from number of laboratories, in which senescent cells were detected in vivo in mice and mouse models. And when these mice were treated with agents which eradicated these senescent cells, numerous signs of rejuvenation were observed.
I’m talking about the first paper of that kind appeared in 2011, Mayo Clinic, and the group led by Jim Kirkland and Jan van Deursen and a series of follow-up papers with similar results. In general, the idea of putting senescent cells in the position of the key sources of sterile chronic inflammation associated with aging came from Judy Campisi, who has provided the most important discoveries in that field.
Well, this theory is extremely appealing for many reasons. First, it is very well supported by evidence. Indeed, senescent cells, when they turn into senescents in culture, switch their phenotype into, so-called, SASPs, and that’s an associated secretory phenotype, the state in which cells continuously secrete pro-inflammatory factors. Second, these cells appear in culture as a result of serial passaging resembling aging. And, therefore, this link became kind of natural between aging and senescent cells. The presumption was that certain cells in the body who used up the number of divisions they can go through before they reach this state may be increasing with age and, therefore, these cells accumulate.
Each of them may become the source of sterile inflammation. Each single one provides a very weak signal, but, when they accumulate altogether, the impact may become significant and translated into pathological conditions. So recently, there were very few – and, even now, it is like that – very few biomarkers of senescent cells, none of which is very reliable because every single biomarker is kind of promiscuous and is not universally selective for senescent cells.
Among these biomarkers, two have been most popular. One is high level of expression of, so-called, senescent-associated senescent-associated beta-galactosidase, which can be detected chemically in fixed cells and tissues which undergo staining, including X-gal, which turn beta-galactosidase reaction into the blue dye under conditions which is not optimal for endogenous beta-galactosidases mammalian cells at low pH. And, under these conditions, the background beta-gal activity of normal cells is practically not seen and senescent cells become brightly visible. So this reaction, which unfortunately requires a cell… It can not be done on paraffin-embedded sections and require preservation of the enzymatic activity and, therefore, is available, mostly, on the frozen sections or in cells in culture… has been used very, very frequently. And in many papers, it has been just the only assay which was used for detection of so-called senescent cells.
Figure 1.Induction of p16Ink4a and SAβG in macrophages does not require p53.
The other biomarker, which resulted from a detailed analysis of promoters which are active selectively in senescent cells is the gene encoding cyclin-dependent kinase inhibitor p16. And the genes name is INK4a. In fact, this promoter of this gene is frequently upregulated in senescent cells, and it has relatively low background in other cells of the organism.
Again, p16 activation is not limited to senescent cells and, moreover, not every senescent cells has elevated p16, but that’s the best we have as of today. That is why, whenever the investigators want to create a mouse model in which they could have the desirable gene expressed exclusively in senescent cells, they use p16 promoter. And there are several mouse models; I’m aware of three in which reported constructs were put under p16 promoter. And the claim was that, when these reporters become obviously expressed in mouse tissues, that was interpreted as accumulation of senescent cells. Also, one can put under this promoter the gene which enables selective eradication of cells with this expression, and, therefore, there is an opportunity to selectively kill such cells. Again, this can be interpreted as a selective eradication of senescent cells.
Using these models, two groups of investigators claim that eradication of senescent cells in aged mice led to substantial demonstration of signs of rejuvenation and, in one case, with increased lifespan. Well, obviously, these data not only provided a very powerful support for the theory about the role of senescent cells in aging but also provided the proof of concept for development of pharmacological approaches to anti-aging treatment and treatment of conditions which lead to the high risk of development of age-related diseases, including cancer.
We obtained such mice in our laboratory, and we have been working with them during last couple of years. The mice we are using are coming from the laboratory of Norman Sharpless from North Carolina. And they have a luciferase reporter gene, which is substituting one of the alleles of p16 and, thereby, being expressed from the p16 promoter. We were pleased to see that these mice accumulate p16-driven luciferase-positive cells detected by in vivo imaging during their lives, which, actually, very well fit the senescent cell theory in their accumulation during life.
However, we were very surprised not seeing accumulation of these cells following total-body radiation or treatment with other genotoxic conditions, which, supposedly, should create lots of senescent cells. We also were puzzled that we were unable to see activation of p16-driven luciferase when we take tissues from these mice and isolated mesenchymal cells from these tissues in vitro and then turn them into senescents, and we failed to see activation of luciferase.
Again, all this together stimulated us to look at the nature of p16-positive cells in these mice and determined their nature, their origin, and their fate in vivo. We started from following the consequences of injection of cells, which would turn into senescents in vitro following injection in vivo into mice. And we labeled cells. We made cell senescents in culture by gum radiation. Then, we injected them intraperitoneal, subcutaneously, into mice. And we looked for their presence by monitoring the label which they were marked with.
Well, it appears that these labeled cells – their traces are disappearing quite quickly, and, within a few days, there are none left in the mice. However, if you put normal cells of similar origin, they actually last much longer. That was the first indication that there may be a mechanism of selective eradication of senescent cells in the body. To check this mechanism and one of our hypotheses was that this mechanism is associated with physical attack of some cells of immunity against senescent cells, and there’s supposed to be innate immunity because it’s happening immediately without any education over the organism.
Figure 5.Poly(I:C) abrogates elevated p16Ink4a expression in two independent in vivo models.
We use a trick in which we embedded senescent cells created in vitro into algenate beads, small spheres consisting of a polymer, which enables to keep cells alive inside them, does not interfere with acquisition of nutrients and oxygen by the cells, but prevents any attack against the cells from any immunocytes. When we took these beads filled with senescent cells and put them in peritoneal cavity of mice, we were pleased to see that they are lasting four weeks without significant death, indicating that senescent cells, who disappear if they are injected without protective beads, are indeed killed by some, so far, unknown mechanism.
In order to identify the executors of senescent cells, we put these beads filled with senescent cells as bait inside, very peritoneal cavity of normal mice, and two weeks later, we pulled them out and analyzed who was accumulating in terms of how cells around these beads in lavage liquid, as well as in the capsule, which was formed around every bead.
Our results brought us to a very important and quite striking observational. The major part of the cells, which was so in these beads as well as in the lavage, appeared to be cells with macrophageal markers on them, which appeared to be bright fluorescence, meaning that they have activated p16, and also positive for beta-gal staining conducted under conditions we are using to reveal senescent cells. So we had to conclude that senescent cells put in the beads attract, probably, by the products of their secretion special subtypes of immune cells, significant proportion of which become reprogrammed to start expressing two biomarkers which people have been using to distinguish senescent cells.
We studied these macrophages in detail, and, after we published our first paper in which we describe this phenomenon, we published a second one, also in Aging, where their properties were described in further details. And we confident that these are bonafide macrophages, not only because they have have biomarkers, they have surface antigen specific for macrophages, but also they are capable of phagocytosis and, moreover, they can be selectively killed by liposome-embedded clodronate, a poison which only kills cells capable of phagocytosis. This killing could be done both in vitro and in vivo when you inject liposomal clodronate inside mice.
So, as far as the presence of these cells in the body of those mice which are not embedded with algenate beads with senescent cells, today, we are confident that these macrophages are accumulating in subcutaneous fat of aged mice in large numbers. And, again, they express biomarkers of macrophages that can be selectively eradicated by clodronate.
So, altogether, it means that the cells which become p16-positive vivo, not necessarily our senescent cells – our operations does not disprove that the signal which we and other investigators are seeing in these mice and increasing with age is not associated with senescent cells. So, potentially, certain proportion of cells we see are, indeed, senescent. However, we are confident that significant part of the signal goes from macrophages, which can be induced into the phenotype associated with expression of both senescent markers when they’re exposed to senescent cells. What is also interesting that this phenotype is reversible. And, in our second paper, we provide a number of physiological stimuli which can either stimulate or suppressed acquisition of this phenotype by macrophages.
All this, together, provides a very interesting step forward in evolution of the theory of aging associated with accumulation of certain specific cell types, contributing to the sterile inflammation occurring in tissues. Today, we can say that those cells which we claim to be the main source of that are not necessarily senescent, but also can be immunocytes who share with senescent cells some of their properties but are not senescent by nature and simply reprogrammed macrophages.
What is the relative impact of these macrophages versus senescent cells towards the process of aging is a very important question, not only from a theoretical standpoint, but also from practical standpoint because, from the time when senescent cells were claimed to be the key players of aging, there have been a substantial effort in the field in generating and testing senolytic compounds, drugs, emerging drugs, which potentially can have anti-aging effect due to eradication of senescent cells from the body.
Whether senolytic compounds would, indeed, solve the issue because, presumably, they will eliminate only a part of the p16-positive cells. To what extent, we need to redirect our attention to the senescent cell-associated macrophages as potential alternative source of secreted factors is an open question. And these are the questions which we are trying to address in our ongoing work, which stems from these observations. Thank you.
Click here to read the full study published in Aging.
—
Aging is an open-access journal that publishes research papers monthly in all fields of aging research and other topics. These papers are available to read at no cost to readers on Aging-us.com. Open-access journals offer information that has the potential to benefit our societies from the inside out and may be shared with friends, neighbors, colleagues, and other researchers, far and wide.