“Hyperfunctional signaling directly drives age-related diseases.”
— Mikhail Blagosklonny, M.D., Ph.D.
BUFFALO, NY- May 18, 2022 – Dr. Mikhail Blagosklonny published his new review paper in Aging (Aging-US) Volume 14, Issue 9, entitled, “Hallmarks of cancer and hallmarks of aging.”
In this review, Dr. Blagosklonny expands on Gems and de Magalhães’ notion that canonic hallmarks of aging are superficial imitations of the hallmarks of cancer. He takes their work a step further and proposes the hallmarks of cancer and aging based on a hierarchical principle and the hyperfunction theory.
“Here I present the hallmarks of cancer, depicted as a circle by Hanahan and Weinberg [1], not as the circle but hierarchically, from molecular levels to the organism (Figure 1).”
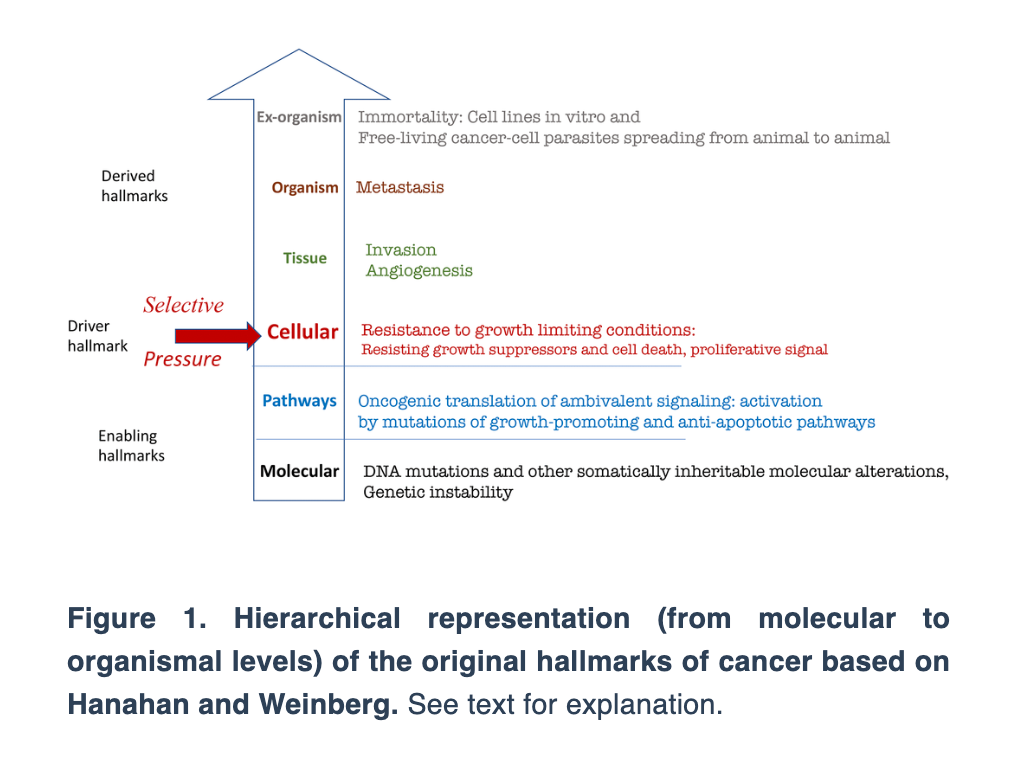
Next, Dr. Blagosklonny depicts the hallmarks of aging suggested by López-Otín et al. based on the hierarchical principle.
“This representation renders hallmarks tangible but reveals three shortcomings (Figure 2).”

The first shortcoming that Dr. Blagosklonny notes is the lack of hallmarks on the organismal level. The second is that the relationship between hallmarks on different levels is unclear. The third is that the inclusion of genetic instability as a hallmark is based on the theory that aging is caused by the accumulation of molecular damage.
“The molecular damage theory was refuted by key experiments, as discussed in detail [44–51].”
Dr. Blagosklonny then uses the hyperfunction theory to arrange the hierarchical hallmarks of aging.
“Let us depict hallmarks of aging, according to the hyperfunction theory of aging (Figure 3).”
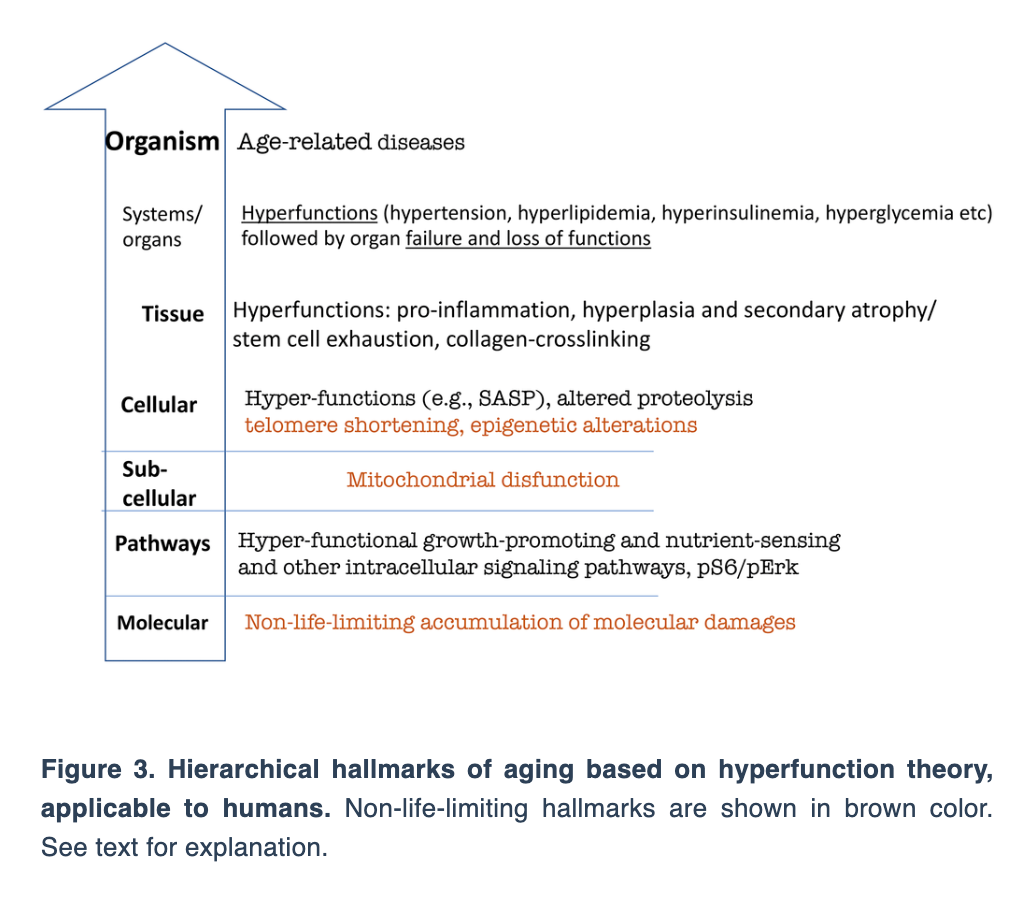
Dr. Blagosklonny continues by discussing the key to understanding aging and aging as a selective force for cancer. He concludes this review by discussing the common hallmarks of cancer, aging and cell senescence.
“In organismal aging, cancer and cellular senescence, the same key signaling pathways, such as mTOR, are involved. This is why the same drugs, such as rapamycin, can suppress all of them.”
DOI: https://doi.org/10.18632/aging.204082
Correspondence to: Mikhail V. Blagosklonny
Email: [email protected], [email protected]
Keywords: oncology, carcinogenesis, geroscience, mTOR, rapamycin, hyperfunction theory
Follow Dr. Blagosklonny on Twitter: https://twitter.com/Blagosklonny

AGING (AGING-US) VIDEOS: YouTube | LabTube | Aging-US.com
About Aging-US:
Launched in 2009, Aging-US publishes papers of general interest and biological significance in all fields of aging research and age-related diseases, including cancer—and now, with a special focus on COVID-19 vulnerability as an age-dependent syndrome. Topics in Aging-US go beyond traditional gerontology, including, but not limited to, cellular and molecular biology, human age-related diseases, pathology in model organisms, signal transduction pathways (e.g., p53, sirtuins, and PI-3K/AKT/mTOR, among others), and approaches to modulating these signaling pathways.
Follow Aging on social media:
- SoundCloud – https://soundcloud.com/Aging-Us
- Facebook – https://www.facebook.com/AgingUS/
- Twitter – https://twitter.com/AgingJrnl
- Instagram – https://www.instagram.com/agingjrnl/
- YouTube – https://www.youtube.com/agingus
- LinkedIn – https://www.linkedin.com/company/aging/
- Pinterest – https://www.pinterest.com/AgingUS/
For media inquiries, please contact [email protected].
Aging (Aging-US) Journal Office
6666 E. Quaker Str., Suite 1B
Orchard Park, NY 14127
Phone: 1-800-922-0957, option 1




